Lesson 6
Key Sentences

| onoːdə̌r xədə̌n be? | What is the date today? | |
| onoːdə̌r ɑrbə̌n sɑr iːn gʊʧə̌n nəg næː odə̌r | Today is October 31. | |
| onoːdə̌r gærə̌g iːn dorbə̌n biʃəː oʧə̌gdə̌r gærə̌g iːn dorbə̌n | Today isn’t Thursday. Yesterday was Thursday. | |
| œrœː tɑː juː xiːx be? | What are you going to do this evening? | |
| tɑnæː torsə̌n odə̌r ʧin xədə̌n sɑr iːn xədnæː odə̌r be? | What month and day is your birthday? | |
| bid ud iːn omə̌n tədnæːd ɔʧjɔː bɔln ʊː? | Let’s go to their place in the morning, okay? |
Dialogs
One

A: onoːdə̌r xədə̌n be?
B: onoːdə̌r ɑrbə̌n sɑr iːn gʊʧə̌n nəg næː odə̌r
A: onoːdə̌r gærə̌g iːn dorbə̌n uː?
B: onoːdə̌r gærə̌g iːn dorbə̌n biʃəː oʧə̌gdə̌r gærə̌g iːn dorbə̌n mɑrgɑːʃ gærə̌g iːn ʤʊrgɑːn
A: œrœː tɑː juː xiːx be?
B: biː sulʤəːnt gɑrnɑː tɑː?
A: biː təlwə̌s uʤnəː
Two

A: tɑnæː torsə̌n odə̌r ʧin xədə̌n sɑr iːn xədnæː odə̌r be?
B: gurbə̌n sɑr iːn ɑrbə̌n dɔlɔː næː odə̌r tɑː?
A: tɑbə̌n sɑr iːn jus næː odə̌r
B: dorbə̌nt ʧiʧig iːn torsə̌n odə̌r
A: dorə̌b næː odə̌r gærə̌g iːn xədə̌n be?
B: gærə̌g iːn odə̌r
A: tɑː tədnæːd ɔʧn ʊː?
B: ɔʧə̌n tɑː?
A: biː bɑs ɔʧnɔː
B: bid ud iːn omə̌n ɔʧjɔː bɔln ʊː?
A: bɔlnɔː
| Notes |
| /xədə̌n/ (or /xədnæː/ or /xəd/) is the word for “how many”. It is used with questions about numbers. |
| Like the months, the days of the week are easy in Mongolian. Monday is “week + 1”, Tuesday is “week + 2”, and so on. Sunday is the exception. It can be said “week + 7” but is usually said “week + day”. |
Substitution
One
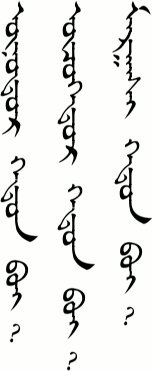
_________ xədə̌n be?
|
Two

_________ xədnæː odə̌r be?
|
Three

A: œrœː tɑː juː xiːx be?
B: biː _________
|
Four

bid _________ bɔln ʊː?
|
| Notes: |
| As you can see in Substitution 3, the /-n/ verb ending can be used to say what you are going to do in the future. |
| The /-jɔː/ ending (or /-jɑ, -jə, -ju, etc. as the case may be) can be used to make a suggestion, as in “Let’s do something.” We can see this in Substitution 4. |
Expansion
One

A: mɑrgɑːʃ xədə̌n sɑr iːn xədnæː odə̌r gɑrə̌g iːn xədə̌n be?
B: mɑrgɑːʃ ɑrbə̌n nəgə̌n sɑr iːn xœrə̌n næːm næː odə̌r gærə̌g iːn odə̌r
Two

miniː næːʤ iːn torsə̌n odə̌r ən gærə̌g iːn tɑbə̌n tər ən jil xœrə̌n nɑs tæː ud iːn xœːn gər təːn uʤə̌x ər ɔʧnɔː
| Notes: |
| /ər/ is another grammar word. Let’s not worry about it now. It will come up again later. (Besides, I don’t understand it enough yet to explain it well anyway. ) |
Vocabulary
| xədə̌n | how many | |
| gærə̌g | week | |
| gærə̌g iːn nəgə̌n | Monday | |
| gærə̌g iːn xɔjə̌rə̌n | Tuesday | |
| gærə̌g iːn gʊrbə̌n | Wednesday | |
| gærə̌g iːn dorbə̌n | Thursday | |
| gærə̌g iːn tɑbə̌n | Friday | |
| gærə̌g iːn ʤʊrgɑːn | Saturday | |
| gærə̌g iːn odə̌r | Sunday | |
| oʧə̌gdə̌r | yesterday | |
| œrœː | evening | |
| juː | what | |
| xiː- | to do | |
| torsə̌n odə̌r | birthday | |
| ud iːn omə̌n | morning | |
| ud iːn xœːn | afternoon | |
| tədnæːd | their place (house) | |
| bɔl- | to be okay | |
| sulʤəːnt gɑr- | get online | |
| təlwə̌s | TV | |
| uʤ- | see, watch, look, visit | |
| ʧiʧigə | QIqige (a common girl’s name meaning “flower”) | |
| nɔm | book | |
| ʊŋʃ- | read | |
| dʊː | song | |
| sɔnə̌s- | listen | |
| ʤæxtə̌l | letter | |
| biʧ- | write | |
| ærxə̌n bɑːr | bar, tavern | |
| nɑstæː | age, years old, old | |
| ər | grammar particle |
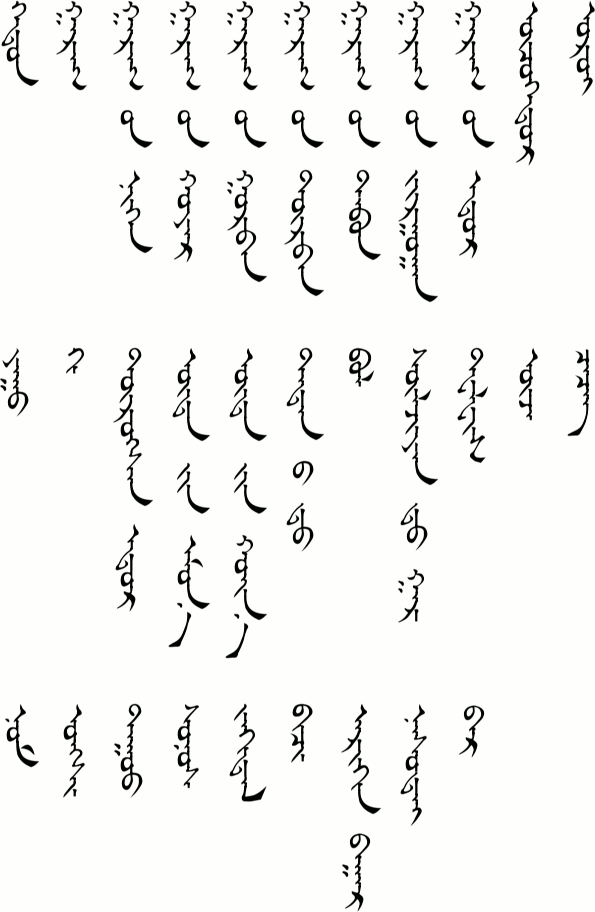
Grammar
Word Order
By now you have probably already noticed that the verbs come last in the sentence. If the verb has an object then the object goes in front of the verb. If we were to use the same order in English then we would get sentences like
- I food eat.
- I book read.
- I letter write.
- I friend visit.
Of course, in English this is poor grammar, but in Mongolian it is perfectly fine. In fact, to use the English grammatical order in Mongolian would be be wrong. They say that Koreans and Japanese can learn Mongolian fairly easily because their grammar is similar. For native English speakers it doesn’t feel natural at first, but with practice we can get used to it, too.
See Substitution 3 above for examples of this word order.
Practice
How do you say the following words:
- how many
- what
- days of the week
- yesterday, today, tomorrow
- morning, afternoon, evening
- birthday
- age
- do
- read
- watch
- listen
- visit
- book
- song
- letter
How do you say the following sentences:
- What day (of the week) is today?
- What day (of the month) was yesterday?
- Tomorrow is Friday.
- When is your birthday?
- What are you doing tonight?
- Let’s watch TV, okay?
- Are you going to their place?
- What day is Sunday?
- Answer the above questions.
Review by saying the following:
- Let me give an introduction.
- How are your parents?
- I’m going to the internet cafe tomorrow.
- Hello.
- Are your parents busy?
- Where are you going?
- Goodbye.
If you were able to say most of these things correctly then you are ready to go on to lesson seven. If not, then keep practicing! You can also download the audio for the whole lesson so that you can practice listening while going to school. Just right-click the link below and choose “Save Link As…” to download.
Lesson 6
![]()
If you have any questions about this lesson or if you notice a mistake, then please leave a comment below. If I don’t know the answer myself, then I will ask our teachers.
I know I keep asking alot of questions but that is a way to learn. I was wondering since the verbs go at the end of the sentence why did bol go before the another word in the sentence a couple of lessons ago, as in “Bi bagsh bishe, bi bol suregsh”. Thanks.
Questions are good. If you have a question about something then other people do, too. And I get to learn a little more by trying to answer it.
I had to look this one up in my grammar book. I guess /bɔl/ isn’t technically considered a verb. It is a connecting particle (or Focus Particle, as my grammar book says.). It connects the subject of the sentence with its complement. It functions similarly to the English linking verb “to be”. However, unlike “is, am, are”, you can’t conjugate /bɔl/. (There is a similar verb /bɔl-/ (to become) and that one can be conjugated at the end of the sentence.)
Thank you so much! That makes alot more sense. Also in the vocabulary list it has bɔl meaning to be okay. How do you tell the difference between that and bɔl to become?
The verb /bɔl-/ can mean “okay” or “to become” depending on the context. It is a commonly used verb with a few different meanings. I forgot that in our lessons so far it has only meant “okay”.
Hi, thanks for your effort on this online course, I really learned a lot!
Just 1 question, when we say “to read a book”, why say “nɔm ʊŋʃə̌n” instead of “nɔmig ʊŋʃə̌n”? I thought nɔm should be the object here.
As I understand it, when talking about reading a book in general you say “nɔm ʊŋʃə̌n” but if you were talking about a specific book, like this book, you’d say “ən nɔmig ʊŋʃə̌n”.